Self-Supervised Learning of Molecular Representations from Millions of Tandem Mass Spectra Using DreaMS

Introduction to Molecular Discovery
Life, at its most fundamental level, is made up of molecules. From DNA to proteins, from vitamins to synthetic drugs, molecules form the structural and functional foundation of all living systems. Despite this central role, scientists have only identified a fraction—less than 10%—of the natural molecules that exist on Earth. This means that an enormous chemical universe remains unexplored, holding the keys to groundbreaking discoveries in medicine, nutrition, materials science, and beyond.
A recent research paper titled “Self-Supervised Learning of Molecular Representations from Millions of Tandem Mass Spectra Using DreaMS” takes a bold step toward navigating this vast molecular space. By combining artificial intelligence with mass spectrometry, the study proposes a method for deciphering the “chemical dark matter” that has long eluded scientific interpretation.
Understanding Natural Molecules and the Challenge of Identification
Natural molecules play critical roles in biological processes such as metabolism, immune response, and tissue repair. Identifying them is essential for understanding life and harnessing nature’s chemistry for applications like drug discovery and diagnostics.
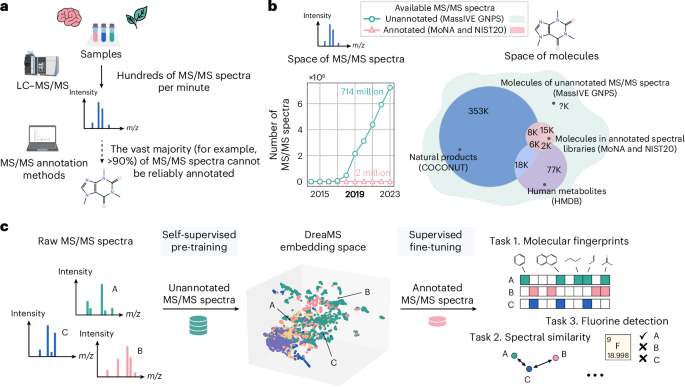
Scientists use a technique called liquid chromatography tandem mass spectrometry (LC-MS/MS) to analyze these molecules. This method separates and fragments samples, generating complex spectra that serve as molecular fingerprints. However, here’s the problem: over 90% of the spectral data remains unmatched to any known molecule, rendering it effectively invisible to current databases.
AI Meets Chemistry: Introducing DreaMS
To tackle this challenge, researchers developed an AI model called DreaMS (Deep Representations of MS/MS Spectra). At its core, DreaMS is a self-supervised neural network designed to interpret millions of unlabeled spectra without needing prior annotations.
Just as language models like GPT learn to understand and generate human language by analyzing massive text corpora, DreaMS learns molecular patterns by examining over 201 million mass spectra. Through this process, the model internalizes the underlying structure and chemical properties represented by the spectra, without ever seeing the actual molecules during training.
The DreaMS Atlas: A Map of Molecular Space
Once trained, DreaMS constructs a multi-dimensional embedding space known as the DreaMS Atlas. Here’s how it works:
- Spectra corresponding to structurally or functionally similar molecules are grouped closely together.
- Dissimilar molecules are placed further apart.
- The map provides a coherent, high-dimensional organization of both known and unknown molecules.
This is akin to how language models cluster synonyms and related words, enabling contextual understanding. In the DreaMS Atlas, molecules that have never been formally identified can now be studied based on their relationships to known compounds.
Applications and Breakthrough Discoveries
The potential applications of DreaMS are far-reaching:
- Taxonomic classification from food spectra: The model could classify food items based purely on their molecular spectra, aligning them with biological taxonomy without needing any label.
- Health insights: The atlas uncovered associations between certain unidentified molecules and specific health conditions, opening new avenues for biochemical and clinical research.
- Hypothesis generation: Researchers can now make educated guesses about unknown molecules’ functions based on their proximity to well-characterized ones.
Fine-Tuning for Targeted Molecular Insights
Beyond classification, DreaMS can be fine-tuned to predict specific chemical features or drug-likeness properties. For instance:
- Drug discovery: Using Lipinski’s Rule of Five, the model can flag molecules likely to be suitable for pharmaceutical development.
- Fluorine detection: Fluorine-containing compounds are highly valued for their stability. DreaMS can now predict fluorine presence with high accuracy.
- Structure prediction: The long-term goal is to enable DreaMS to reconstruct full molecular structures from spectral data—turning it into a true AI-powered discovery engine.
Open Source and Community Collaboration
One of the most exciting aspects of this project is its open-source philosophy. The developers have made the DreaMS model and training code available on GitHub and HuggingFace, complete with instructions for mapping spectra and fine-tuning the model for new applications.
This transparency invites collaboration from the broader scientific and machine learning communities. Whether you’re a chemist, a data scientist, or a biotech entrepreneur, DreaMS offers a framework you can build upon to accelerate molecular discovery.
Conclusion: A New Era of Molecular Understanding
DreaMS represents a paradigm shift in how we explore the molecular world. By combining self-supervised learning with mass spectrometry, it unlocks a treasure trove of previously inaccessible chemical knowledge. As researchers continue to refine the model and extend its capabilities, we stand at the threshold of a new era in molecular discovery—one where machines help us chart the vast and largely uncharted chemical space that underpins life itself.
References
- Bushuiev, R., Bushuiev, A., Samusevich, R., Brungs, C., Sivic, J., & Pluskal, T. (2025). Self-supervised learning of molecular representations from millions of tandem mass spectra using DreaMS. Nature Biotechnology. https://www.nature.com/articles/s41587-025-02663-3
- Pluskal, T., et al. (2025). Self-supervised learning of molecular representations from millions of tandem mass spectra using DreaMS. ChemRxiv. https://chemrxiv.org/engage/chemrxiv/article-details/67f52eee6dde43c908f23b84
- IDSL_MINT: a deep learning framework to predict molecular fingerprints from mass spectra. Journal of Cheminformatics. https://jcheminf.biomedcentral.com/articles/10.1186/s13321-024-00804-5
- MassFormer: Tandem Mass Spectrum Prediction for Small Molecules using Graph Transformers. https://arxiv.org/abs/2111.04824
- MSBERT: Embedding Tandem Mass Spectra into Chemically Rational Space by Mask Learning and Contrastive Learning. Analytical Chemistry. https://pubs.acs.org/doi/10.1021/acs.analchem.4c02426
Future Work
The DreaMS framework opens several avenues for future research and development:
- Integration with Other Analytical Techniques: Combining DreaMS with other analytical methods, such as nuclear magnetic resonance (NMR) spectroscopy, could provide a more comprehensive understanding of molecular structures.
- Real-Time Analysis: Implementing DreaMS in real-time analytical settings could facilitate immediate identification and characterization of compounds during experiments.
- Expansion of Training Datasets: Incorporating more diverse and comprehensive datasets into the training process could enhance the model’s ability to identify.